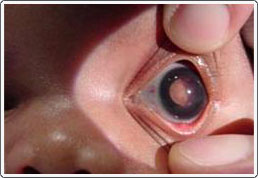
Newborn with retinoblastoma, left eye. Note the whitish shape seen through the pupil. This is a tumor in the back of the eye.
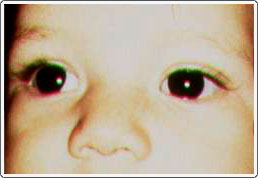
Child with exotropia and slight hypertropia of the right eye caused by a retinoblastoma tumor in that eye. Note the location of light reflexes on the pupils.
Retinoblastoma is a malignant tumor of the sensory retina and is the most common ocular malignancy in childhood. Critical to the treatment of retinoblastoma is early identification, as cure rates are over 90% if the tumor is localized within the eye. A white reflex within the pupil (leukocoria) is a serious sign that could suggest retinoblastoma and that deserves immediate referral. If untreated, seedling nodules produce secondary tumors that gradually fill the eye and extend along the optic nerve to the brain, resulting in death. Although retinoblastoma is rare (estimated at 1:20,000 live births), it must be considered in any child with leukocoria.
Newborn with retinoblastoma, left eye. Note the whitish shape seen through the pupil. This is a tumor in the back of the eye.
Child with exotropia and slight hypertropia of the right eye caused by a retinoblastoma tumor in that eye. Note the location of light reflexes on the pupils.
Clinical Signs
The most common presenting signs of retinoblastoma are strabismus and a white pupil. Retinoblastoma may also present with findings that mimic other ophthalmic disorders such as primary angle-closure glaucoma, vitreous hemorrhage, retinal detachment, hyphema, hypopyon, and even preseptal cellulitis. Usually, retinoblastoma presents with a quiet eye; however, the presence of the tumor can lead to problems such as intraocular hemorrhage (bleeding in the eye), inflammation (swelling of the eye and eye tissue) and a red, painful eye.
Genetics of Retinoblastoma
Retinoblastoma is caused by a mutation in a certain gene found on chromosome 13q14. The mother and father must both possess this mutated allele to have a retinoblastoma tumor occur in a child. There are two recognized types: sporadic retinoblastoma and hereditary retinoblastoma.
Sporadic Retinoblastoma
The sporadic form of retinoblastoma is caused by a spontaneous mutation in both parents’ cells at the time of conception. This requires two independent mutational events that are not inherited. Approximately 60% of all retinoblastoma cases are the non-hereditary (sporadic form). The tumor is primarily unilateral, and the eye usually has one main tumor.
Hereditary Retinoblastoma
Patients with hereditary retinoblastoma have one defective retinoblastoma gene in virtually all cells in their body. Because of this, patients with hereditary retinoblastoma are predisposed to acquiring secondary non-ocular tumors in late childhood and adulthood. Most inherited retinoblastomas are bilateral, with only 15 % being unilateral. Hereditary retinoblastoma is often multifocal, with multiple tumors developing in each eye. Patients with the hereditary form present early, around 12 months of age on the average. The majority of new cases of bilateral hereditary retinoblastoma are due to a new mutation, as only 10% of affected patients have a positive family history. If a parent has retinoblastoma, there is a 50 % chance of passing the predisposition to their child. If there is no family history of retinoblastoma and a child is born with bilateral retinoblastoma, the chance that another sibling will develop retinoblastoma is approximately 8%. With DNA testing, carriers of a retinoblastoma gene mutation can be identified in most cases.
Treatment of Retinoblastoma
If the tumor is bilateral, then every attempt should be made to save at least one eye. Large tumors involving the macula are associated with a poor visual prognosis and are generally treated by removal of the affected eye (enucleation). Smaller tumors can be treated with external beam radiation, most useful for posteriorly located tumors. Radioactive plaque treatment, which involves attaching a radioactive “button” directly over the site of the tumor, has also been used and has the advantage of minimizing radiation effects on normal tissue. External beam radiation may have the disadvantage of inducing or speeding up the development of secondary tumors in patients with the hereditary form of retinoblastoma. The use of chemotherapy to decrease the size of the tumor, followed by laser or cryotherapy, has been effective in eyes with some vision. Small peripheral tumors can be treated with cryotherapy or laser photocoagulation.
An infant who presents with unilateral retinoblastoma must be followed closely, since 20% will develop a new tumor in the “good eye.” This risk diminishes greatly after two years of age. In cases of hereditary retinoblastoma, siblings following are at risk for developing this tumor. They should be followed with serial retinal examinations unless DNA analysis demonstrates the child does not carry a retinoblastoma gene.
Prognosis of Retinoblastoma
Unilateral retinoblastoma that has been treated with enucleation, and is without extension into other parts of the eye, is associated with long-term survival of the patient in over 90% of cases. If the tumor cells extend quite posteriorly, the survival rate lowers to approximately 60%.. External beam radiation performed in patients without extrascleral extension results in a cure for approximately 85% of patients. Successfully treated retinoblastoma patients have a risk period for extraocular spread of tumor between 1 to 2 years.
If the patient has hereditary retinoblastoma, he/she is at risk for developing secondary malignant tumors later in life. These tumors are often in the area of the external beam radiation, but can also occur in remote sites. Patients who have had radiation therapy develop secondary tumors earlier than patients who have not had radiation. The incidence for developing a secondary tumor in patients with hereditary retinoblastoma is probably over 50% with a long-term follow-up of over 30 years.
